Doença de Caroli: causa de dor abdominal recorrente na infância
Ulysses Fagundes-Neto, Sérgio Thomas Schettini, Jamal Wehba, José Pinus, Francy Reis da Silva Patrício
Fonte: J Pediatria 54(4):154-158, 1983
Caroli (1), em 1958, descreveu pela primeira vez uma síndrome rara caracterizada por dilatação sacular segmentar dos ductos biliares intra-hepáticos. Posteriormente, Ker & cols. (2) introduziram o termo fibrose hepática congênita para caracterizar uma entidade clínica onde fibrose portal formando verdadeiros micro-hamartomas múltiplos constituíam a lesão fundamental. A partir de então tem sido verificada uma associação frequente entre doença de Caroli e fibrose hepática congênita. Atualmente admite-se que ambas as entidades clínicas constituem diferentes estágios de uma mesma enfermidade (3). Assim sendo, são reconhecidas quatro formas de apresentação a saber (4): fibrose periportal, colangítica, mista e latente.
A forma pura da enfermidade que se manifesta clinicamente com colangite e formação de litíase biliar, constitui-se, no entanto no padrão mais incomumente descrito, sendo conhecidos aproximadamente apenas 20 casos da doença de Caroli na forma pura de apresentação. A doença cística intra-hepática é também frequentemente associada a dilatação congênita dos túbulos renais (rim medular esponjoso). Esta afecção renal é usualmente destituída de significado clínico e deve ser diferenciada da lesão renal do tipo rins policísticos, a qual pode ser grave e que se associa à doença hepática policística (6).
Neste trabalho descrevemos dois casos da doença de Caroli em crianças em idade escolar com manifestação clínica de dor abdominal e crises intermitentes de icterícia.
RELATO DOS CASOS
Caso 1 – E.A.D., 11 anos, sexo feminino foi encaminhada ao Serviço de Cirurgia Pediátrica da Escola Paulista de Medicina com queixa de crises de dor abdominal e icterícia com colúria há um ano. Nesta ocasião suspeitou-se de hepatite aguda; a icterícia cedeu espontaneamente ao cabo de duas semanas, mas as dores abdominais persistiram esporadicamente. Há cinco dias voltou a apresentar dores abdominais em cólica localizadas no hipocôndrio direito, suficientemente intensas para obrigar a menor a suspender todas as suas atividades. Como antecedentes pessoais foi referido que as crises de cólica se iniciaram por volta do segundo ano de vida. Nesta época esteve internada em outro Serviço durante 34 dias tendo sido submetida a pelo menos duas intervenções cirúrgicas para extirpar inúmeros “tumores abdominais” (sic), que, segundo informação transmitida ao pai da criança, eram de natureza benigna. A partir de então a menor esteve novamente internada por três vezes, devido a crises de dor abdominal e icterícia com diagnóstico de hepatite crônica. Na presente internação, ao exame físico a menor encontrava-se em bom estado geral, afebril, anictérica. Apresentava cicatriz cirúrgica na região do hipocôndrio esquerdo e o fígado era palpável a 3 cm do rebordo costal com contornos nítidos, regulares e consistência normal. Foi internada com suspeitas diagnósticas de hepatite crônica, colecistite crônica calculosa e doença de Caroli. Foram realizados os seguintes exames laboratoriais: hemoglobina: 12.4 g%, hemossedimentação: 40 mm, bilirrubina total: 0.56 mg%, direta: 0.15 mg%, indireta: 0.41mg%, transaminases: TGO 219 mU/ml (VN 0-12), TGP 220 mU/ml (VN 0-12), atividade de protrombina: 76%, eletroforese de proteínas: albumina3.9g%, alta1 0,14g%, alfa2 0.91g%, beta 1,12g%, gama 1,33g%. Colangiografia intravenosa mostrou exclusão da vesícula além das vias biliares intra e extra-hepáticas.
No 11ₒ dia de internação a paciente foi submetida a laparotomia para realização de biópsia hepática a céu aberto e exploração radiológica das vias biliares. 0 aspectoₒ macroscópico revelou fígado aumentado de tamanho, endurecido, vinhoso, finamente granuloso, sem nodulações grosseiras ou tumorações. Foi obtido fragmento de tecido hepático para estudo anatomopatológico que revelou fígado com arquitetura preservada e ductos biliares aumentados em número e tamanho, contendo por vezes, neutrófilos em seu interior. Hepatócitos com presença de pigmento biliar no citoplasma, por vezes apresentando aspecto degenerativo focal e proliferação conjuntiva bem evidente alargando algumas áreas portais. Em conclusão, quadro histológico de colangite crônica recidivante. Colangiografia pelo cístico evidenciou árvore biliar dilatada intra e extra-hepática, especialmente dilatações caliciformes intra-hepáticas compatíveis com as características anatômicas descritas na doença de Caroli (Fig. 1).
Durante o ato cirúrgico foram realizadas: colecistectomia, duodenostomia, papilotomia, coledocotomia e drenagem das vias biliares a Catell; foi também efetuada drenagem do leito vesicular e da área duodenal abordada com dreno e penrose que foi exteriorizado por contra-abertura da parede abdominal.
0 pós-operatório transcorreu sem intercorrências e a paciente recebeu alta hospitalar no 11ₒ dia após a cirurgia para seguimento ambulatorial. Transcorridos 18 meses da intervenção cirúrgica, a paciente encontra-se sob controle, em bom estado geral, apesar de esporadicamente apresentar crises de dor abdominal.
Caso 2- A.P.B., 7 anos e seis meses, sexo feminino que apresenta queixa de crises recorrentes de dor abdominal, as quais se iniciaram há 18 meses. Por ocasião do primeiro episódio de dor abdominal houve aparecimento de icterícia associada a colúria com descoramento das fezes. Nesta época foram solicitados os seguintes exames da função hepática: TGO 70mU/ml, TGP 60mU/ml (VN 0-12), bilirrubinas (total: 6.0mg%, direta: 2.5mg%, indireta: 3.5mg%), eletroforese de proteínas (PT: 7.6g%, albumina 3.6g%, alfa1 0.16g%, alfa2 1.15 g%, beta 1.28g%, gama 1.40g%), antígeno HBs negativo. Hepatite viral aguda foi o diagnóstico considerado. A paciente foi acompanhada ambulatorialmente. A icterícia cedeu rapidamente e as transaminases voltaram aos valores normais após dois meses (bilirrubina total: 0.8mg%, indireta: 0.4mg%, TGO: 28, TGP: 20). As crises de dor abdominal voltaram a se repetir por três vezes mais. A dor abdominal era de intensidade suficientemente intensa para impedir a paciente de exercer suas atividades normais, e o aparecimento da mesma não guardava relação com qualquer situação específica, tendo surgido inclusive durante momentos em que a paciente estava brincando, sendo obrigada a suspender a brincadeira. Em outras ocasiões a crise dolorosa surgia à noite quando a menor estava dormindo, sendo a mesma despertada pela dor. Os episódios de dor abdominal evoluíam por quatro a cinco dias cedendo espontaneamente ao cabo desse período de tempo. Cada crise dolorosa durava cerca de uma hora, e era resistente a terapêutica antiespasmódica. Após o episódio doloroso, a menor voltava a desempenhar suas atividades normalmente.
Na época da terceira crise de dor abdominal a menor esteve internada durante um mês com a finalidade de investigar mais profundamente a etiologia do processo em causa. Foram realizados os seguintes exames: Hb 12.3g%, hemossedimentação 5mm, urina tipo I normal, reação sorológica para toxoplasmose negativa, prova de falcização das hemácias negativa, pesquisa de porfirobilinogênio na urina negativa, transaminases: TGO 300 UFR (VN 8-40), TGP 350 UFR (VN 5-30), gamaglutamiltranspeptidase 246 mU/ml (VN 4-18), bilirrubinas totais: 1.30mg%, direta 0.70mg%, indireta 0.60mg%, eletroforese de proteínas: PT 6.0g%, albumina 3.48g%, alfa1 0.24g%, alfa2 0.66g%, beta 0.60g%, gama 1.02g%. Ultrassonografia hepática inconclusiva, revelando excesso de zonas sonolucentes que podiam corresponder a imagem das veias supra-hepáticas ou colestase intra-hepática. Foi realizada biópsia hepática de agulha por punção intercostal que revelou arquitetura hepática com estrutura preservada, e discreto alargamento dos espaços portais. Hepatócitos mostravam-se tumefeitos com citoplasma granuloso e núcleos ovoides. Sinusóides com congestão e células de Kupffer discretamente hipertrofiadas. A paciente apresentou remissão espontânea das crises dolorosas tendo recebido alta sem esclarecimento diagnóstico.
A paciente permaneceu assintomática pelo espaço de um ano, mas há dois dias voltou a apresentar queixa de dor abdominal intensa e febre, sendo encaminhada ao Setor de Gastrenterologia Pediátrica da Escola Paulista de Medicina. Ao exame físico encontrava-se em regular estado geral, febril T: 38ºC, evidenciando fácies de sofrimento, prostrada, anictérica, abdome discretamente distendido, doloroso a palpação, mas sem sinais de irritação peritoneal; fígado palpável a 3 cm do rebordo costal com características normais. Foi levantada suspeita de doença de Caroli e solicitada colangiografia intravenosa que mostrou dilatação das vias biliares intra-hepáticas de maior calibre, configurando-se o diagnóstico de doença de Caroli (Fig. 2).
A menor passou a ser acompanhada ambulatorialmente e após 18 meses apresentou evolução satisfatória com crises esporádicas de dor abdominal e febre. Nestas ocasiões a administração de Ampicilina e anti-espasmódicos tem sido eficaz no controle das crises de colangite.
Discussão
Apesar de ser considerada uma afecção bastante rara, a incidência real da doença de Caroli deve ser maior do que a referida na literatura, pois mais de 50% dos pacientes portadores de cisto de colédoco (afecção de embriogênese estreitamente relacionada a entidade em questão) parecem ter em associação algum grau de dilatação biliar intra-hepática tipo Caroli (7). A associação parece ser ainda mais frequente nas dilatações cilíndricas e não císticas da via biliar comum sendo mais frequente também o acometimento do lobo esquerdo do fígado. Caroli (8) relata ainda que na forma pura desta lesão (dilatação policística congênita das vias biliares intra-hepáticas) existe frequentemente associação com moderada dilatação do ducto biliar comum. Por este motivo tem sido considerada como um subtipo ou variante anatômica de um grupo de afecções consideradas globalmente como “cisto de colédoco” (tipo IV) (9).
A despeito da doença de Caroli ser reconhecidamente uma entidade clínica de natureza congênita (10) a maioria das descrições na literatura dizem respeito a adultos jovens. A manifestação de dor abdominal recorrente na infância é extremamente frequente constituindo-se numa síndrome sintomatológica de difícil abordagem diagnóstica (11). Dor abdominal recorrente é geralmente traduzida por uma queixa vaga e subjetiva permanecendo sem esclarecimento na grande maioria dos casos (12). Torna-se importante, porém, distinguir dor abdominal recorrente de provável origem não orgânica daqueles casos aonde a dor é provocada por algum substrato patológico bem definido. De acordo com Galler e cols. (13) a frequência de uma enfermidade orgânica diagnosticável como causa de dor abdominal recorrente na infância depende de vários fatores, incluindo a idade da criança, a presença de outros sintomas gastrointestinais e a região geográfica do mundo aonde o levantamento é realizado. No caso dos pacientes aqui descritos duas informações clínicas adicionais permitem levantar forte suspeita da dor referida ser devida a alguma causa orgânica. Em primeiro lugar a associação de icterícia em ambas as pacientes e em segundo a localização da dor no abdome afastando-se da região periumbilical. Mais ainda, o fato de ambas as crianças serem despertadas do sono por crise dolorosa e também serem obrigadas a interromper suas brincadeiras. Todos estes dados segundo Apley (14) são fortemente sugestivos da existência de lesão orgânica como causa da dor abdominal. Nas nossas pacientes embora não fosse possível comprovar a existência de cálculo biliar, a dor abdominal é facilmente explicável pelas crises de colangite caracterizadas pela presença de febre e icterícia. Em uma delas inclusive a colangite foi comprovada pela biópsia hepática a céu aberto. A confirmação diagnóstica é preferencialmente realizada por meio da ultrassonografia (15), colangiografia retrógrada ou por colangiografia transhepática ou ainda pela colangiografia intra-operatória (16. 17) . Uma de nossas pacientes teve o diagnóstico definido por colangiografia intra-operatória, enquanto que a outra por não haver indicação cirúrgica imediata foi submetida como primeira tentativa diagnóstica a colangiografia intravenosa. Esta revelou-se, na nossa opinião, suficientemente esclarecedora para não deixar margem a dúvidas quanto ao diagnóstico de doença de Caroli. Assim sendo, não consideramos necessária a realização de qualquer outro procedimento técnico.
A feitura de uma derivação bilio-digestiva seria benéfica apenas para diminuir o componente de estase de origem extra-hepática, ou seja, aquela devida ao cisto de colédoco. A derivação bilio-digestiva não iria beneficiar, no entanto, de maneira significativa as condições de drenagem da árvore biliar intra-hepática dilatada.
lnfelizmente, na doença de Caroli o diagnóstico precoce não está associado a um prognóstico mais favorável. 0 surgimento das crises de colangite e o consequente risco de septicemia é inevitável (16). Tem sido referida também associação com carcinoma em 2.5% dos casos (18). Nossas pacientes vêm apresentando evolução temporária satisfatória e durante as crises de colangite são administrados antibióticos com o objetivo de superar o processo infeccioso vigente.
Imagens
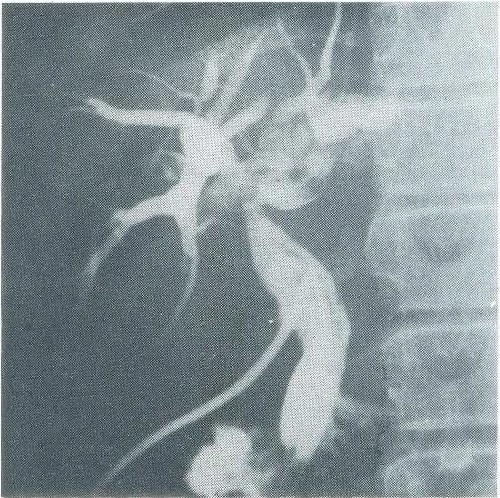
Figura 1 – Colangiografia retrógrada intra-operatória evidenciando dilatação dos ductos biliares intra-hepáticos.
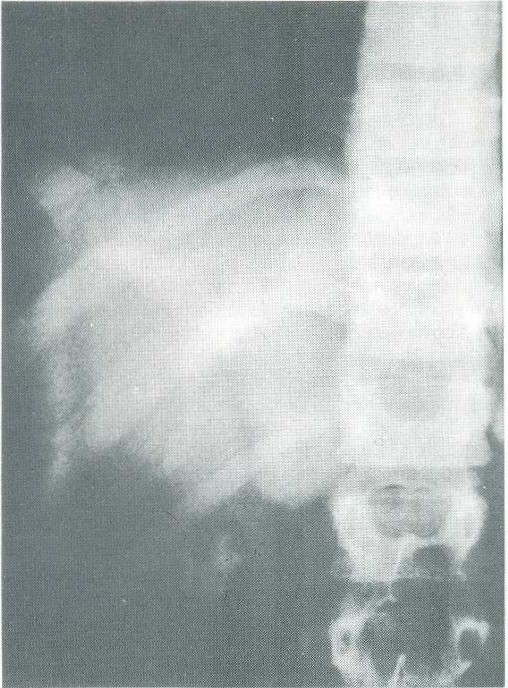
Figura 2 – Colanqiografia intravenosa mostrando dilatação cística dos ductos biliares intra-hepáticos
REFERÊNCIAS BIBLIOGRÁFICAS
- CAROLI, J; SOUPAULT, R; KOSSAKOWSKI, J; PLOCKER, L; PARADOWSKA, M: La dilatation polykystique congenitali des voies biliares intra hepatiques. Essai de classification. Sem. Hasp. Paris, 34: 488, 1958.
- KERR, DNS.; HARRISON, CV; SHERLOCK, S et al.: Congenital hepatic fibrosis. Q. J. Med., 30: 91-117,1961.
- DUSOL, M; LEVI, JU; GLASSER, K; SCHIEFF, ER: Congenital hepatic fibrosis with dilation of intrahepatic bile ducts. Gastroenterology, 71:839-843, 1976.
- FAUVERT, R; BENHAMOU, JP; MEYER, P: Fibrose hepatique congenitale. Fr Etud Clin. Biol., 9, 375-377, 1964.
- PRIDGEN, JR; AUST, B; MciNNIS, DW: Primary lntrahepatic gallstones. Arch Surg, 112: 1037-1043, 1977.
- FOULK, W L: Congenital malformations of the intrahepatic biliary tree in the adult. Gastroenterology, 58· 253-256, 1970.
- TSUCHIDA, Y; ISHIDA, M: Dilatation of the intrahepatic bile ducts 1n congenital cystic dilatation of the common bile duct. Surgery, 69· 776-781. 1971.
- CAROLI, J: Diseases of the intrahepatic biliary tree. Clin. Gastroenterol, 2: 148-153, 1973.
- FLANIGAN, DP: Biliary cysts. Ann. Surg. 182: 635-643, 1975.
- LONGMIRE, WP; MANDIOLA, SA; GORDON, HE: Congenital cystic disease of the liver and biliary system. Ann. Surg, 174: 711-726, 1971.
- APLEY, J: The child with abdominal pains. London: Blackwell Sctentific Publications. 1959.
- BATY, BM: Abdominal pain in childhood. J. Iowa Med. Soc, 55: 681, 1965.
- GALLER, JR; NEUSETEIN, S; WALKER, WA: Clinical aspects of recurrent abdominal pain in children. Year Book Medical Publishers. Inc, 1980. p. 31-53.
- PRINGLE, MLK; BUTLER, NR; DAVIE, R: 11,000 Seven Year Olds. London: Longmans. 1966.
- BASS, EM; FUNSTON, MR; SHAFF, M.I: Caroli’s disease an Ultrasonic diagnosis. Brint. J. Radiol., 50:366-368. 1977.
- HERMANSEN, MC; STARSHAK, RJ; WERLIN, SL: Caroli disease: the diagnosis approach. Pediatr., 94: 879-882, 1979.
- ROSEWARNE, MD: Cystic dilatation of the intrahepatic bile ducts. Brit. J Radiol. 45: 825-827,1972.
- JONES, AW; SHREEVE, DR: Congenital dilatation of intrahepatic biliary ducts with cholangiocarcinoma. Brit. Med. J., 2: 277-278, 1970.